
极端压缩条件下氢的结构与相图理论模拟研究当前位置:首页 > 科学技术 > 研究进展
极端压缩条件下氢的结构与相图理论模拟研究
文章来源:科技信息中心编辑室 时间:2018-11-12 访问数:
共1页 1
在高压物理领域,基于量子力学的数值模拟其核心在于求解电子和原子的多体薛定谔方程,并获得相应温度-压力下的自由能。基于平均场近似的密度泛函理论和Kohn-Sham方程,为解决多电子体系的量子问题提供了既便利而又足够精确的方法,但对于核的量子效应却无能为力。由于原子核的量子效应在氢及其化合物中具有举足轻重的作用,为了准确模拟轻核的量子效应和非谐振动,我们提出了路径积分的多层抽样算法,并独立开发了第一原理路径积分分子动力学程序。分层抽样的基本思想是将原子间的相互作用分拆成长程、中程和短程等部分,再利用实空间路径积分的离散特点,将它们分别赋予不同的路径积分离散点,长程部分利用精确的第一原理和少量的离散点计算,而中程和短程部分则利用不同精度的有效原子间相互作用势和更精细的路径积分离散点得到。我们证明了:在同样的收敛条件下,此方法可以在不损失计算精度的前提下大幅提高第一性原理路径积分的效率,而有效原子间相互作用势只影响计算效率但不对最终的路径积分结果造成影响。在此基础上,我们还开发了包含非谐声子贡献的完整自由能计算方法和液体结构的各向异性分析技术。这些方法和技术,结合电子部分的密度泛函计算和原子结构的统计分析,可模拟极端压缩条件下氢及其化合物的结构变化与奇异量子行为,所获得的部分代表性结果简述如下。
1 发现金属氢新的结构和简并基态
利用第一性原理计算方法和结构搜索与预测技术,系统研究了高压下高密度氢的结构稳定性,发现金属氢接近基态的低能结构远比预先想象的复杂。在400 GPa压力以下,我们计算的结果与先前的理论预估吻合很好:C2/c相在100 GPa时成为基态,在270 GPa左右转变到Cmca-12相。Cmca则在370 GPa时变得较稳定,但很快便与Cmca-12形成简并。然而与之前的认识不同,我们的计算除了确认原子相的金属态Cs-Ⅳ在490 GPa压力下变得稳定外,还发现了与之简并的Fddd相。其他的简单原子相例如BCC、FCC、HCP以及它们的各种低对称变形结构相在2 TPa压力之前都是不稳定的。
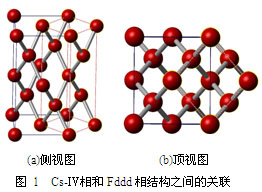
图1给出了在能量上完全简并的高密度金属氢基态Cs-Ⅳ相和Fddd间的几何结构关系,在每个Fddd相的晶胞中包含两个Cs-Ⅳ相的晶胞,其中黑实线标出了Fddd的晶胞,红色短划线给出了变形的Cs-Ⅳ相的晶胞结构。二者都可从立方金刚石结构导出:对金刚石结构的立方胞进行四角变形(增加c/a比)即可得到Cs-Ⅳ相,对后者再进行正交变形(增加b/a比)则可获得Fddd结构。结构畸变改变了原子周围的最近邻粒子分布。因此,这两个结构间虽然存在很紧密的几何对应关系,但它们的局部原子环境不完全一样。其中,Fddd中氢原子有着更小的最近邻原子间距(小于0.1 nm),因此Fddd相可能有更强的“残余”化学相互作用,即还有部分氢具有双原子H2分子的属性。
在更高的压力区域,结构搜索方法预测其基态将具有H3的特征团簇。然而既不同于早期预测的R3m或R-3m结构,也不同于后来预测的Cmcm结构,我们发现在有限温度下稳定的相结构事实上为C2221,如图2所示。该结构也具有H3特征团簇,它是Cmcm的低对称变形。Cmcm虽然具有与C2221几乎一样的焓值,但当考虑原子核的动力学效应后,在分子动力学模拟中其在20 K的低温下自发转变到C2221结构。分析发现Cmcm结构更紧凑一些,但二者的最近邻都有12个原子,已经很接近金属中常见的BCC、FCC、HCP等相的配位数。其电子结构也呈现出类似金属的清晰的近自由电子特征,但同时在费米面附近有明显的p电子占据,并形成sp杂化,这可能是该结构稳定性的根源。此外,由于H3键很弱,不足以抗拒原子的热运动,因此该特征团簇有可能只是瞬态的。
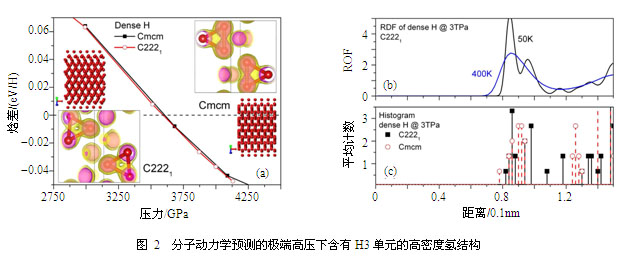
上述研究结果说明:高密度氢从氢分子离解开始的多种相互作用竞争在2~3 TPa时已经基本结束,高密度氢已逐渐转变为简单金属。
2 首次确定高密度氢固相间的势垒
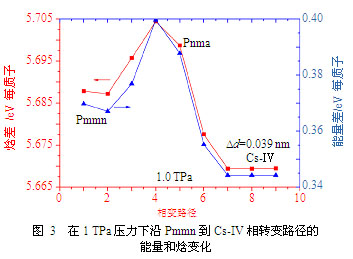
势能曲面形状和分隔各相的势垒高低对理解高压下氢的行为十分关键。我们利用自行改进的NEB方法,计算了1 TPa压力下从亚稳的Pmmn相到基态Cs-Ⅳ相的相变路径及相应的能量和焓的变化,如图3所示。其中的势垒以Cs-Ⅳ相为基准,焓垒大约为0.035 eV/H,能垒约为0.055 eV/H。两个相沿相变路径的能量曲面都有一个比较平缓的区域。特别是基态Cs-Ⅳ结构,其在理想结构附近的能量/焓的变化非常小,暗示在该结构中允许氢原子发生最大约Dd=0.039 nm左右的原子漂移,与此形成对比的是该相中原子间最近邻距离为0.092 nm。这一发现意味着高密度氢(不管是基态还是亚稳结构)中存在相当宽广而平坦的能量低谷,而平坦的势能曲面将导致远比准谐振近似小得多的零点能。此外,由于从Cs-Ⅳ到Pmmn的焓垒0.035 eV/H在能量上大约相当于400 K,意味着在这一压力范围高密度氢在室温下仍处于固态。当然,考虑到CS-Ⅳ相宽广平坦的能量曲面,其熔化温度在零点振动的驱动下进一步降低也是有可能的。
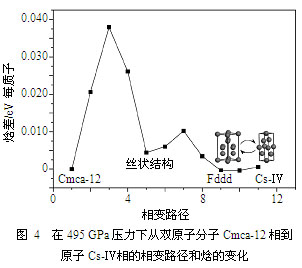
我们还研究了495 GPa压力下高密度氢从双原子分子Cmca-12相离解到单原子的Cs-Ⅳ相的相变路径和势垒,结果如图4所示。我们得到了一个高度约为DH=0.038 eV/H的焓垒。势垒的存在必然导致相变的滞后并在低温下推高表观的离解压力。由于Cmca-12与Cs-Ⅳ的原子体积差约为DV=3′ 10-5 nm3,因此在一阶近似下可能的压力提升为DP=DH/DV?200 GPa。也就是说,如果考虑势垒的动力学影响,密度泛函预测的双原子分子氢的离解压力应该在700 GPa左右。其他可能的影响还包括:(1)质子的零点运动,这有利于高对称的单原子相,因而将降低离解压力,但其精确的数值大小目前很难预估;(2)密度泛函自身的误差,由于其偏向于均匀分布的电子密度,从而使得计算得到的离解压力被低估了大约50 GPa;(3)存在其他焓垒更低的相变路径,我们不能完全排除这种可能性,因为NEB本质上是一个局域优化算法,所能搜索的相空间范围有限,因此其结果在一定程度上依赖于所选取的初始路径。综合考虑所有这些因素之后,离解压力应该小于750 GPa(或550 GPa,如果不考虑势垒导致的延滞效应的话)。图4揭示的另一个有趣的现象是在简并的基态Cs-Ⅳ和Fddd相之间没有观测到势垒的存在,换句话说,高密度氢有可能在零点运动的驱使下在这两个相之间振荡。注意虽然Fddd可以被看作为Cs-Ⅳ的一种畸变,从晶体学的观点看它其实是金刚石结构的正交变形,即高密度氢基态结构的量子振荡可能远比图4所预示的要复杂,然而在后续的分子动力学模拟中我们并没有观测到这种振荡发生,显示它极有可能是一种稀有事件。此外,该图所示的相变路径上还出现了另一个各向异性且低对称的“丝状”结构,该结构与先前理论预测的氢的低对称结构十分相似,它与其他相间有势垒存在,因而极可能是亚稳的。
3 确定金属氢的零压回收问题
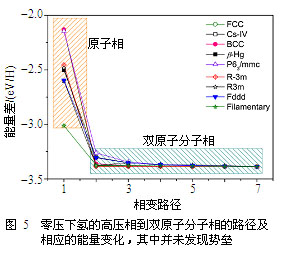
我们用(体积可变的)NEB方法研究了零压下从这些高密度相到双原子分子结构的相变路径及相应的势垒。高密度相的初始结构通过谨慎地(外加约束条件)从极端高压弛豫到零压来获得,而双原子分子结构则通过扰动/弛豫相应的高密度结构获得。典型的结果如图5所示,结论是令人沮丧的:迄今所知的所有低能结构,无论是各向同性的单原子相还是低配位数的三原子分子或链状分子结构,它们在零压下都是不稳定的,即它们与基态的双原子分子结构之间没有任何势垒存在。特别地,早期预言的“丝状”结构也不例外。目前没有发现任何物理机制可以阻止这些高压相自发地蜕变到双原子分子结构。这一物理事实源于低压下氢强烈的配对倾向,并意味着回收氢的高密度相有可能是“非常困难的”。尽管我们的结果是在密度泛函的PBE近似下获得的,这一构筑于均匀电子气模型的局域或半局域交换-关联能量密度泛函倾向于高估金属相的稳定性,并引入了一定的数值偏差,但还不至于将势垒完全抹除(如果严格解中存在势垒的话)。而且,对DFT本身的误差分析显示如果目前的DFT(包含有单电子近似等处理)没有探测到势垒的话,那么即使在更严格的多体量子理论层面,这些已知的氢的高密度相也极有可能是不可回收的。
4 高压下金属氢固体相的稳定性问题
利用两相法模拟,我们发现在1.5 TPa压力下Fddd结构的高密度氢在50 K温度以上首先转变到类似液体但却各向异性的特殊相(有可能是玻璃态,但在较低温度下有明显的长程关联),后者在250 K左右发生熔化进入均匀的各向同性液相区。这一结果与我们对熔化温度的预估是相符的,但却不能告诉我们在极低温度下固体相与液体相之间直接的竞争关系如何。为了回答在绝对零度下处于基态的高密度氢是液体还是固体这一基本问题,原则上我们需要通过比较自由能的大小来确定哪一个相更为稳定。自由能计算是很困难且需要大量的计算资源的。我们因此利用在50 K温度时高密度氢中准谐振近似和非谐振声子的内能均已经收敛到零点能,且零点能与自由能的偏差小于10-5 eV/H这一事实,通过路径积分分子动力学计算50 K时内能及焓的大小来确定趋于基态时各相的相对稳定性。注意虽然准谐振近似对高密度氢来说是一个比较粗糙的近似,但它给出的内能和自由能的量级大小却是在合理的范围之内,因此这一方法是合理可靠的。
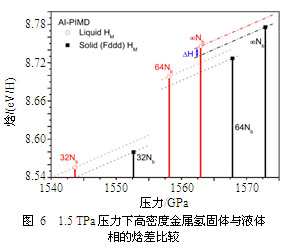
焓的第一原理路径积分分子动力学计算分别利用了32和64个离散点来模拟积分路径,并外推至无限个离散点的极限情况。图6给出了液态和Fddd相的焓的比较,由图可见在热力学的意义上固体相比液体要更为稳定一些。在32个离散点的计算中,二者焓差约为16 meV/H,模拟路径的离散点增加到64个时焓差减小到14.7 meV/H,Richardson外推法给出的极限收敛时的焓差为14.3 meV/H,因此我们可以确定地说核的量子效应并没有给予液态太多的额外优势,在收敛的路径积分条件下固态仍旧要更为稳定。这是“密度泛函精度下高密度氢在1.5 TPa压力附近下的基态是固体”这一基本判断的第一个直接的第一原理数值模拟证据。从我们大量的50 K温度下晶体结构的路径积分分子动力学计算可推知,在更低温度下发生核的波函数自发离域化的可能性几乎没有。在我们所涉及的热力学条件下,固体相的积分路径的弥散程度(近似等于波函数在坐标空间所覆盖的区域大小)主要由势阱约束,而非德布罗意热波长,因此进一步降低温度不大可能会对量子隧穿概率有很大的增强。此外,包含全同粒子的交换作用也很难对熔化温度有定性的改变,因为它对液体自由能的贡献很小(在4He中约为1 K量级),而高密度氢中固、液相的焓差在160 K量级。
5 建立极端高压下氢的基本相图
我们系统研究了高密度氢的熔化曲线与相图,发现高压下氢的熔化曲线存在一个极小值的平台,宽度大于1 TPa。在更高压力下,几乎所有接近基态的低能晶体结构的熔化温度均随压力上升,显现出一个深“U”形的熔化曲线反常形貌。5 TPa以后,高密度氢回归高对称的立方结构,具有自由电子特征,显示这时固态氢已属于Wigner晶体。
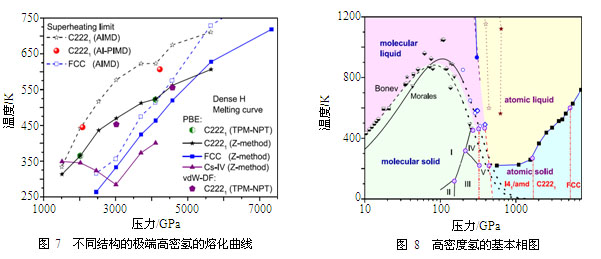
在中低压力下,计算所获得的熔化线在300 GPa附近与Liu等人计算的结果吻合良好,也与Howie等人用DAC高温拉曼光谱测量估计的熔化线的下限相互自洽,这一下降趋势与Kechin熔化方程的外推结果是一致的。然而在H2离解以后,进入原子金属状态的高密度氢的熔化曲线走势发生了急剧变化,首先在500 GPa左右与离解线的交汇附近熔化温度陡然下降,随后进入约250 K的平台区域,这一区域很宽,约有1000 GPa左右。在更高压力下,氢的熔化曲线的走势与之前预期的完全相反,它开始上升,而非下降。我们对目前已知的这一压力区域的所有可能结构,包括最新发现的C2221和高对称的FCC和BCC结构等都进行了检测,发现在压力超过2 TPa后所有这些结构的熔化温度都逐渐升高,这一效应与所用的方法无关(无论Z方法、两相法、AIMD或PI-AIMD模拟),也与所用的电子交换关联泛函无关(广义梯度近似PBE和范德瓦尔斯泛函vdW-DF均给出几乎一样的结果),如图7所示。这确切无疑地显示在足够高压力下高密度氢的熔化曲线具有正斜率,且越来越大。这充分说明Kechin方程的高压外推是不可靠的,从而Ashcroft和Kechin推测的(以及Chen等预测的)高密度氢的熔化温度将趋于绝对零度这一观点的物理根据是不充分的。深U形熔化反常揭示高密度氢更类似于碱金属:即熔化曲线先降低,进入一低谷平台,然后再升高。氢的量子运动(零点运动)使平台附近的熔化温度有100 K左右的下降,但不足以改变熔化线的整体走势和熔化机制。产生深U形低谷的物理根源在于压缩导致的H2分子的离解和s→p电子的激发。这是理论上首次明确这一反常的物理机制,反映了电子结构变化、分子离解与晶体熔化之间复杂的相互耦合关系。它同时还说明在更高压力下不存在高密度氢的量子熔化和量子液体(至少在Wigner晶体熔化之前),因此最可能的量子液体或Ashcroft假设的超流超导体只可能以亚稳的过冷液体(或流动固体)的形式存在于500~2000 GPa,这大大缩小了寻找这一新奇物相的空间范围。
结合DAC拉曼测量和第一性原理的基态稳定结构预测结果和分子动力学模拟结果,我们初步给出了高压下氢的基本相图结构(见图8),这是首次获得如此大范围的相图信息。它清晰地展示了离解线和熔化线极小值之间的直接关系,并暗示了固体H2中结构复杂性的根源。参考实验给出的第四相和第五相的相界,以及计算模拟的熔化线的变化走势,我们推测出了分子相额外的三个三相点位置,以及分子相与原子相间的两个三相点。在原子相的固体区,由于结构存在从Cs-Ⅳ(或者Fddd)向C2221和FCC的转变,因此也有两个三相点存在。这些认识对于理解高密度氢的高压行为,有针对性地发展高压实验技术和能力具有重要意义。
6 新的固-液中间态——流动固体
通常情况下固体和液体具有截然不同的物理性质:固体具有固定形状,可以抗拒外加应力引起的变形,而液体具有流动性,因而没有特定形状(随容器改变而变化)。而且,处于单晶形态的固体具有各向异性,而液体一般是各向同性的。这些性质差异使得固-液相变成为凝聚态物质中物理性质变化最剧烈的相转变之一。正是由于固体和液体的这些性质上的互斥特性,二者间总存在一清晰明确的界面,相应的相变也是一阶的。然而并没有理论能严格证明物理上的熔化必须是一阶的。固-液连续相变有可能存在,但一直没有直接或间接的物理证据。其物理图像来源于林德曼的熔化理论,在这一简单而直观的图像中熔化并不一定必须是一阶的,有可能存在连续的熔化。在现实中介于固-液之间的中间态也确实存在,一个典型例子是液晶,其中质点的运动是类液体的,但分子指向有序排列,具有晶体的部分特征。另一个例子是超离子化合物,其中重离子形成固定形态的晶格,而轻离子像液体一样在其间自由流动。但这两个都不是真正意义上的固-液中间态,理论上提出的第一个同一物质既参与流体运动又参与形成固体晶格的固-液中间态是Andereev和Lifshitz提出的超固体,利用氦超冷固体中量子化的空穴准粒子,他们认为氦有可能在保持固体形态的同时具有(处于玻色-爱因斯坦凝聚状态的空穴导致的)超流体特性。
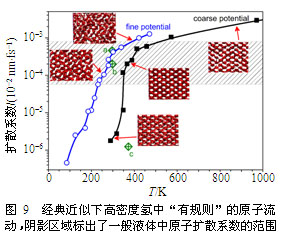
在极端高压下,在氢的深U形反常熔化线的底部平台附近,我们发现高密度氢也具有类似的行为,更为奇特的是这一固-液中间态不需要借助量子的非局域性质而存在(见图9~图10)。导致这一发现的最初线索是我们发现Chen等计算的高密度氢的熔化线事实上并不对应于固体到均匀各向同性液体的熔化,它的终态事实上类似于液晶或玻璃态。因此氢在这一压力状态下存在两个熔化温度,第一个转变温度是从固体到一个具有流动性的各向异性结构的相变,第二个则对应于从该新相到各向同性液体的熔化。我们最初将这一新相假定为玻璃态或无定性态,但随之发现这与很强的原子流动性相矛盾。利用大量精细的路径积分分子动力学模拟,我们发现该特征对粒子量子行为的依赖并不敏感,而且原子的迁移能力可以在(统计平均的意义上)保持原子结构几乎不变的条件下大幅提高(见图10),这暗示着原子分布各向异性和流动性是这一新奇物相的主要特征,即同一物质同时拥有晶体和液体的特性。为了进一步确认这一发现,我们研究了经典情形,发现在去除粒子的量子效应后,原子分布的规律性变得更为明显,并清晰地显示了长程有序的晶体特征和液体流动行为共存(见图9)。通过对结构进行优化,并对原子间的相互作用进行适当近似,我们获得了可以清晰展示这一新相特征的物理模型,进而最终确认它就是处于固-液中间态的新相,我们将之命名为“流动固体”。
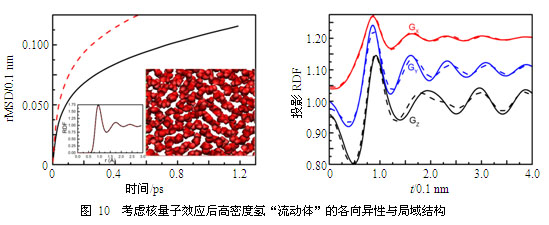
利用此物理模型,我们展示了高密度氢从常规晶体到流动固体的一阶相变,以及伴随的大幅提升近3个量级的原子扩散系数。在流动固体中,原子的迁移能力与液体不相上下,并具有一定的各向异性,其中原子大范围的迁移运动通过分析分子动力学的原子运动轨迹清晰可辨,但它们在长时间、系综统计的意义上是相互关联的,并形成规则的网格结构。这些运动轨迹的长时间系综平均给出了完整有序的原子密度分布,显示了该相所具有的长程有序、各向异性等晶体特征。如果将原子扩散按不同方向投影,分解出的均方位移清晰地展示了原子移动的各向异性特征。从该相向各向同性液体的熔化并不是间断的,而是固体特征逐步消散退化,缓慢过渡的。受限于分子动力学模拟方法本身的局限,我们不能严格证明这一熔化是连续相变,但我们的模拟清晰显示即使这一过程是一阶的,其导致的不连续性必然十分微弱。
流动固体相的发现为理解固液熔化、超固体、以及寻找连续熔化和熔化临界点提供了新的思路和方向。我们的研究结果直观地揭示了液体的流动性和晶体在位形空间的长程有序性可以和谐地出现在量子和经典情形,并指出高密度氢中发现的新颖结构有可能就是流动固体。这一结构中的固-液共存特性暗示当其中的氢原子进入量子状态后它有可能变为超固体,因为高密度氢的流体分量被预测将有超流体转变。这一机制显然不同于Andreev和Lifshitz提出的4He中玻色缺陷子非局域化后导致的宏观量子效应。因此新发现凸显了搜寻高密度氢的量子液态以及极端压缩条件下可能的奇异超固体状态的重要性。
7 轻元素化合物中的新奇结构
富氢化合物是寻找室温超导体的理想舞台。2015年法国CEA的实验研究小组在金刚石压腔中成功合成出了富氢化锂LiHn。令人意外的是他们的实验观测与理论预测大相径庭:他们合成出的富氢化锂是光学透明的晶体,因此是绝缘的、非金属的,而所有理论预测的锂的富氢化物却都是金属。这一理论与实验的严重背离让人困惑不解:一方面实验的数据看起来是可靠的,而另一方面对于氢-锂这一只含s和p电子的主族间离子化合物来说,没有理由怀疑当前的理论存在问题。
为了解决这一谜团,我们详细研究并对比了单质氢和富氢化锂中氢分子的离解过程,发现通常理论所用的密度泛函方法在描述氢分子离解过程中的电子局域化和电荷转移时是有先天缺陷的,需要引入新的修正——即必须考虑范德瓦耳斯相互作用(通常认为这一作用是很弱的,因此在富氢化物研究中常常被忽略)。我们利用新的高精度的方法重新计算了富氢化锂的高压稳定结构和相图,发现范德瓦耳斯相互作用的贡献出乎意料地重要,它甚至完全改变了之前理论预测的基本物性特征:具有金属性质的LiH6相消失了,取而代之的是绝缘的、具有宽带隙(因而是光学透明)的新相LiH9,同时金属性的LiH2也变成了非金属,如图11所示。这一重要的结果消除了实验与理论间的尖锐矛盾,解开了富氢化锂中的谜团。其中问题的关键在于带负电的H3团簇,以前研究人员坚信这一团簇只在重元素的富氢化物中存在,不可能出现在富氢化锂中。但新的研究显示范德瓦耳斯力使得这一奇特的团簇可以稳定存在,且导致了实验观测到的光学透明特性。事实上,如果没有H3团簇,即便考虑了范德瓦耳斯修正,富氢化锂仍将是金属性的。
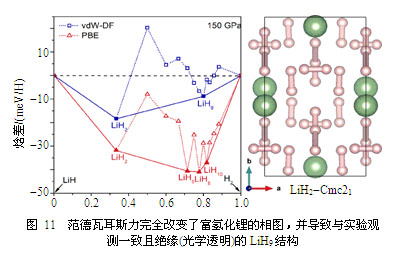
这一新的结果对研究富氢化合物及其金属化机制具有重要意义。研究人员已经从理论上预测了许多的富氢化物,其中一些化合物还被预测具有很高的超导转变温度,但除了极少数(例如H3S)被实验证实之外,其余大部分的理论预测与实验相距甚远。研究人员对其中所涉及的物理过程认识很不充分可能是原因之一。我们的这一进展无疑在此向前迈进了一大步。此外,研究还显示范德瓦耳斯力可使得锂吸附更多的氢,其最高化学配比可高至1∶10,相应的储氢重量比接近60%,遥遥领先于其他的储氢方法或材料,展现了富氢锂化物在新能源应用中诱人的前景。